Glioma is a major tumor type that derives from glial cells of the central nerve system, which is classified from Grade I to Grade IV, with increasing exacerbation according to histology. Large-scale genomic aberration and gene expression studies on gliomas have lead to identifications of genes, which are involved in numerous cellular functions and metabolic pathways, as well as those related to neural development, cell signaling, and tumor suppression. However, the existing literature related to genomic alterations and their molecular mechanisms in gliomas lacks information from Chinese populations. Recently, a study compared genomic aberrations between low-grade and high-grade gliomas (LGG and HGG) in Chinese populations to unveil differences of tumor-related sequence variations and individual genes in the context of pathways and function categories, has been published by Prof. YU Jun and his team at Beijing Institute of Genomics (BIG), Chinese Academy of Sciences (CAS), associated with a research team at First Hospital of Jilin University. Genomic variations between LGG and HGG in Chinese population were mapped based on Illumina’s Beadchip and the results were validated using real-time qPCR. The results have shown that genomes in HGG tend to be deleted more intensively than those in LGG at both cytoband and molecular levels. They have also validated many locations, genes, pathways, and function categories in keeping with the previous studies at different levels. By uncovering molecular differences between LGG and HGG in this study, which are largely based on pathology, morphology, and degree of malignancy, the authors are hoping to associate genetics to clinical outcomes in fighting the disease, including diagnosis, treatment, and prognosis. A network of pathways related to LGG, HGG, and others. (Imaged by Dr. Wang Dapeng) Paper link: http://dx.plos.org/10.1371/journal.pone.0057168 CONTACT: 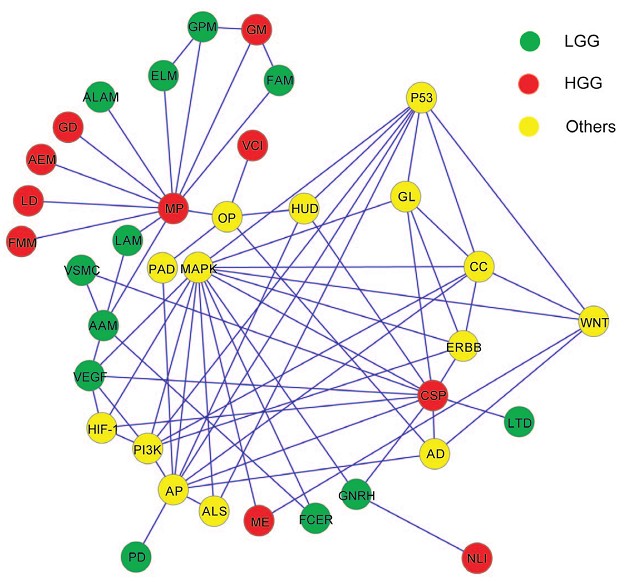
Dr. WANG Dapeng
E-mail: wangdp@big.ac.cn
