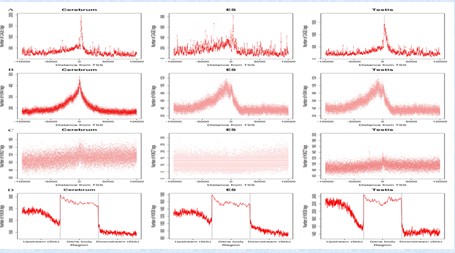
2012-02-07 A discovery of thousands of novel transcripts (mostly non-coding RNAs) that are expressed in mouse cerebrum, testis, and embryonic stem (ES) cells, through an in-depth analysis of rmRNA-seq data was reported by Professor HU Songnian’s group, Beijing Institute of Genomics (BIG), Chinese Academy of Sciences (CAS). The high-throughput next-generation sequencing technologies provide an excellent opportunity for the detection of less-abundance transcripts that may not be identifiable by previously available techniques. These newly detected transcripts show significant associations with transcriptional start and elongation signals, such as the significant enrichment of histone marks (histone H3 lysine 4 trimethylation, H3K4me3), RNAPII binding sites and cap analysis of gene expression tags that mark transcriptional start sites at the upstream of these transcripts. Also, they observed enrichment of histone H3 lysine 36 trimethylation (H3K36me3) along the length of these transcripts. Moreover, these transcripts show strong purifying selection in their genomic loci, exonic sequences, and promoter regions, implying functional constraints on the evolution of these transcripts. These results define a collection of novel transcripts in the mouse genome and indicate their potential functions in the mouse tissues and cells. This research results were published online in frontiers in genetics on December 26, 2011 at http://www.frontiersin.org/non-coding_rna/10.3389/fgene.2011.00093/full The 5′ CAGE and histone modification around novel TU-TSS or gene bodies in the cerebrum, testis, and ES cell. (A) - (D) Profiles of 5′ CAGE, H3K4me3, H3K27me3, and H3K36me3. (Image by LIU Wanfei and ZHAO Yuhui) Contact: Prof. HU Songnian Email: husn@big.ac.cn