GEN: Open-access Data Portal Integrating Transcriptomic Profiles Under Various Conditions
With the development of high-throughput sequencing technologies, RNA sequencing has become a routine and indispensable approach for systematically characterizing transcriptomic profiles across diverse biological conditions at both bulk and single-cell levels. The huge amount of transcriptomic data generated at unprecedentedly exponential rates has posed great challenges in large-scale data aggregation and standardized processing.
To address these challenges, the research group of the National Genomics Data Center, Beijing Institute of Genomics of Chinese Academy of Sciences/ China National Center for Bioinformation (CNCB) developed Gene Expression Nebulas (GEN), an open-access data portal integrating transcriptomic profiles under various conditions, which was published online in Nucleic Acid Research.
In the current version, based on unified data processing pipelines and structured curation model, GEN houses expression profiles of 323 high-quality datasets, including 157 bulk and 166 single-cell datasets. These datasets cover 50,500 samples and 15,540,169 cells across 30 species and are further categorized into 6 biological contexts, namely baseline, genetic, phenotypic, environmental, spatial and temporal.
To provide opportunities for integrative analysis at both transcriptional and post-transcriptional levels, GEN integrates transcriptomic profiles on gene/transcript expression, circular RNA expression, RNA editing and alternative splicing for 10 bulk datasets.
Moreover, GEN also provides abundant gene annotations based on value-added curation of transcriptomic profiles for a total of 1,191,846 genes. Thus, GEN serves as a basic resource for biological and medical researchers to better understand genetic regulatory mechanisms from tissues to cells.
To facilitate large-scale data query, retrieval, analysis and visualization, GEN provides user-friendly web functionalities and applications. Specifically, it delivers online services and offline toolkits for both bulk and single-cell RNA-seq data analysis and visualization, which include differential expression analysis, weighted gene co-expression network analysis, functional enrichment analysis, gene regulatory network inference, cell trajectory inference, cell type annotation, and so on.
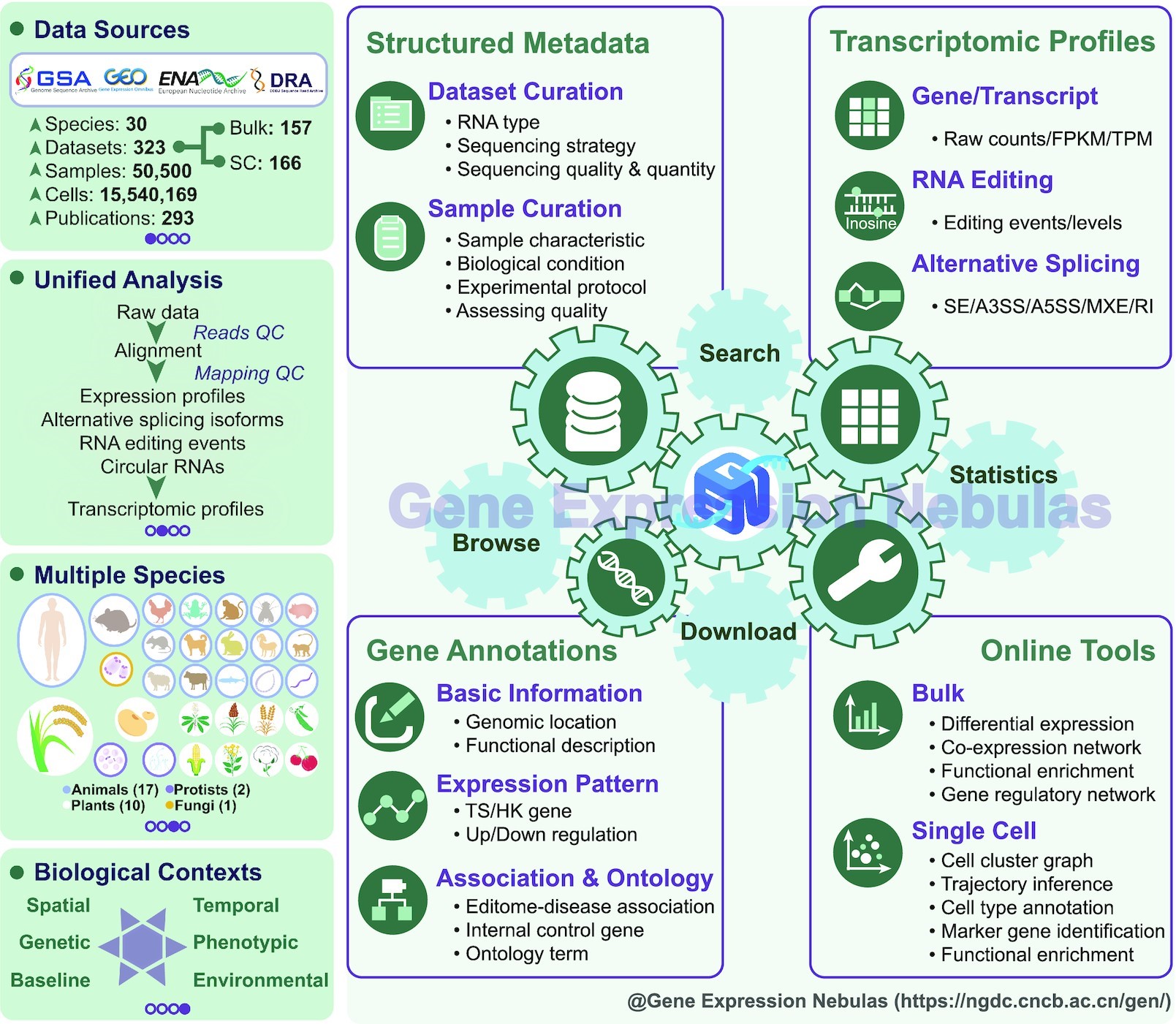
Database contents and features of Gene Expression Nebulas (Image by ZHANG Zhang’s group)
Contact:
Dr. HAO Lili
Email: haolili@big.ac.cn
Dr. ZHANG Zhang
Email: zhangzhang@big.ac.cn