McAN: A novel Computational Algorithm and Platform for Constructing and Visualizing Haplotype Networks
Haplotype networks are graphs for intuitively depicting the evolutionary processes and relationships between sequences, and play an important role in tracking the evolution and migration of different species. In the context of infectious diseases, the sequences of haplotype network nodes contain spatiotemporal information about virus transmission and mutation, making it easier to analyze the dynamic mechanisms of virus transmission and mutation. During the COVID-19 pandemic, several studies used haplotype network methods to analyze the spread and characteristics of the virus in local areas. However, with the explosive growth of SARS-CoV-2 genomic data, existing haplotype network construction algorithms cannot meet the needs of rapid analysis of massive data. Improvements or enhancements to these algorithms are urgently needed.
Recently, the research group of SONG Shuhui at National Genomics Data Center, Beijing Institute of Genomics of Chinese Academy of Sciences (China National Center for Bioinformation) proposed a haplotype network construction algorithm—McAN, which is based on minimum cost arborescence, and developed a haplotype network construction and visualization platform to meet the needs of analyzing massive pathogen genomic data. This work was published online in Briefings in Bioinformatics with the title "McAN: a novel computational algorithm and platform for constructing and visualizing haplotype networks".
McAN extracts four haplotype network construction criteria based on epidemiological characteristics and genetic principles, abstracts the haplotype network construction problem into an integer programming problem according to these criteria, and solves the integer programming problem. The optimality of the McAN algorithm is also proven. Specifically, by reducing the calculation of sequence distances, McAN improves the speed of haplotype network construction. Tests on a data set of about 1,000 whole genome sequences of SARS-CoV-2 show that McAN is two orders of magnitude faster than traditional methods. Tests on a large data set of 5 million SARS-CoV-2 genomic sequences show that the McAN algorithm takes about 25 minutes (50 threads) and has the ability to process massive pathogen genomic sequences. Tests on simulated data sets show that McAN has a memory consumption reduction of over 90% compared to traditional methods without sacrificing accuracy. Additionly, McAN produces reasonable results on several data sets, including monkeypox and influenza A.
Users can obtain the McAN source code from BioCode or GitHub, or construct haplotype networks online and use hierarchical clustering algorithms to partition lineages and view haplotype network results interactively in the self-developed virus haplotype network browser.
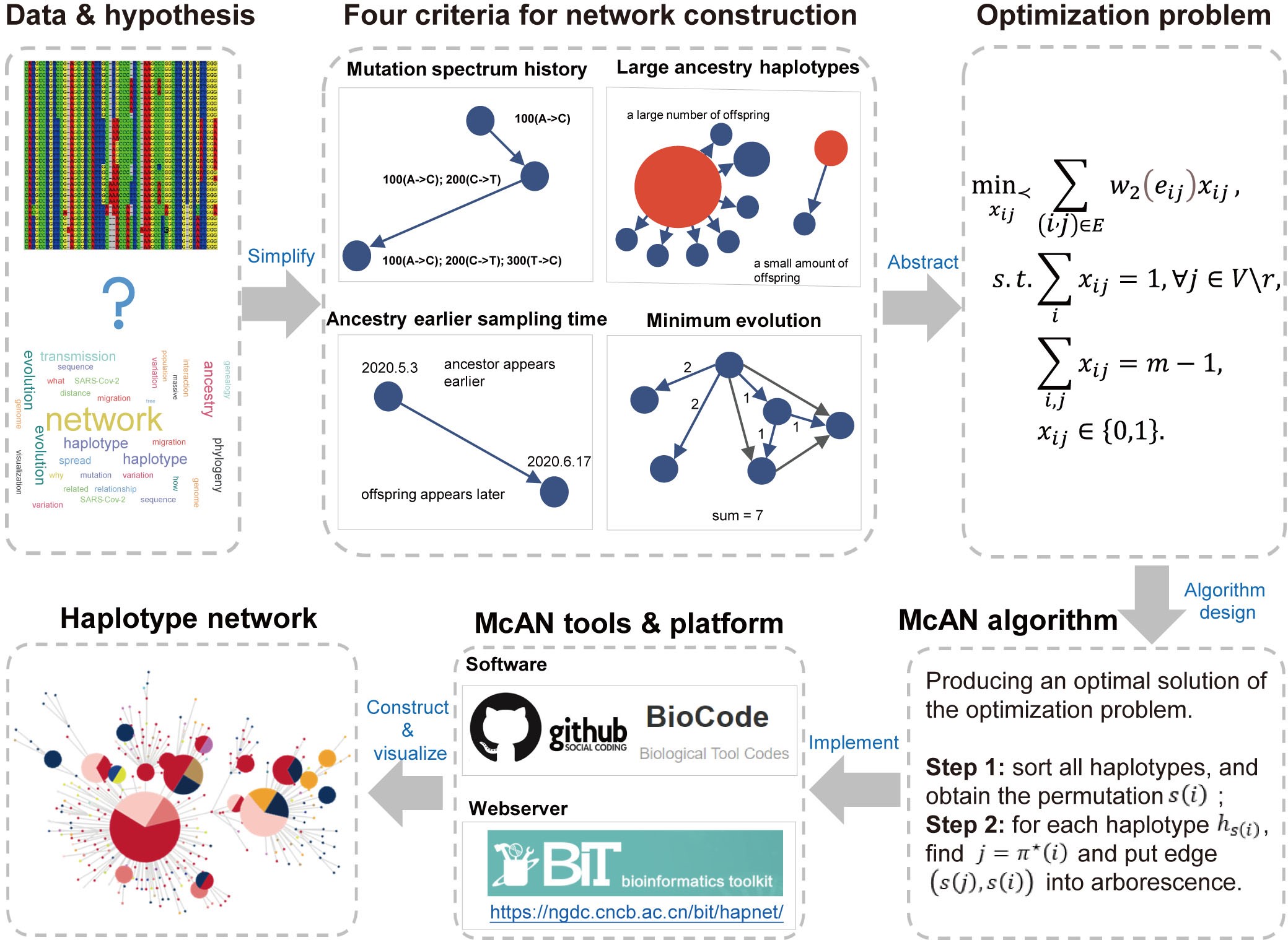
Schematic illustration for haplotype network construction by McAN (Image by National Genomics Data Center)
Contact:
Dr. SONG Shuhui
Email: songshh@big.ac.cn