Researchers Clarify a Role of VGLL3 in DNA Double-strand Break Repair and Chemotherapy
Recently, a collaborated research group led by Profs. GUO CAIXIA from the China National Center for Bioinformation, TANG TIE-SHAN from the Institute of Zoology of the Chinese Academy of Sciences discovered that VGLL3 modulates chemosensitivity through promoting DNA double-strand break repair. This study was published in Science Advances.
DNA double-strand breaks (DSBs) are one of the most deleterious types of DNA lesions, which can result in mutagenic events or cell death if left unrepaired or repaired inappropriately. In general, DSBs are mainly repaired by non-homologous end joining (NHEJ) and homologous recombination (HR). While NHEJ is active throughout interphase to facilitate the direct end ligation in an error-prone fashion, HR is restricted to the S-G2 phases of the cell cycle to allow for error-free repair by using a homologous sister chromatid as a template.
VGLL3 is a member of the VGLL family that contains a conserved TONDU motif (TDU), a region essential for binding with TEA domain-containing transcription factors (TEADs). Distinct from other VGLL family members, VGLL3 also contains a unique glutamate-rich (E-rich) motif at the N-terminus and a histidine-rich (H-rich) motif at the C-terminus. As a candidate female-biased immune regulator, the elevated VGLL3 expression is associated with inflammatory diseases, especially female-biased autoimmune diseases.
Recently, emerging evidence implicated a role of VGLL3 in tumor development. VGLL3 was reported to associate with cancer progression by promoting tumor cell proliferation or motility, with its expression positively correlated with poor prognosis in multiple types of human cancers, including breast, ovarian, colon and gastric cancers. Paradoxically, a tumor suppressor role of VGLL3 was also reported in estrogen receptor (ER) positive breast cancers, in which VGLL3 is essential in mediating Hippo/Yes-associated protein (YAP) signaling to repress ESR1 (encoding ERα) expression. Moreover, VGLL3 was reported to confer therapeutic resistance to TEAD–YAP blockade through functioning as a transcription co-activator to induce the expression of YAP-suppressed genes. Nevertheless, unlike its homolog VGLL4, how VGLL3 functions in tumor development remains largely obscure.
In this study, the researchers report that VGLL3 can be recruited to DSBs in a PARylation- and chromodomain helicase DNA-binding protein 4-dependent manner. Through its E-rich motif, VGLL3 associates with KLHL15, an E3 ubiquitin ligase adaptor, to prevent CtIP from proteasomal degradation. VGLL3 also associates with MDC1 through its H-rich motif to enhance the MDC1-USP7 interaction and to limit the interaction between MDC1 and thyroid hormone receptor interactor 12 (TRIP12), a known E3 ubiquitin ligase for USP7. Depletion of VGLL3 downregulates the expression of CtIP and MDC1 at the protein level, impairing the recruitment of multiple DNA damage response (DDR) factors to DSBs. The unique E-rich and H-rich domains enable VGLL3 specific functions in stabilizing CtIP and MDC1, thereby synergistically promoting HR, which is not conserved among other VGLL family members. As a functional consequence, VGLL3 deficiency impairs HR repair and sensitizes cells to multiple DSB-inducing agents. Consistently, targeting VGLL3 inhibits tumor growth and sensitizes xenografted tumors to etoposide treatment. Together, their data reveal that VGLL3 can function as a DDR regulator that is essential for optimal initial DDR signaling and DSB end resection, which is independent of its roles in transcriptional regulation.
It is highly likely that VGLL3 may also regulate the stability of other DDR factors besides CtIP and MDC1, which remains to be identified in the future studies. Given that DDR can protect tumor cells from genotoxic drug treatment, a better therapeutic outcome can be achieved by inhibition of VGLL3 DDR function and its association with TEAD.
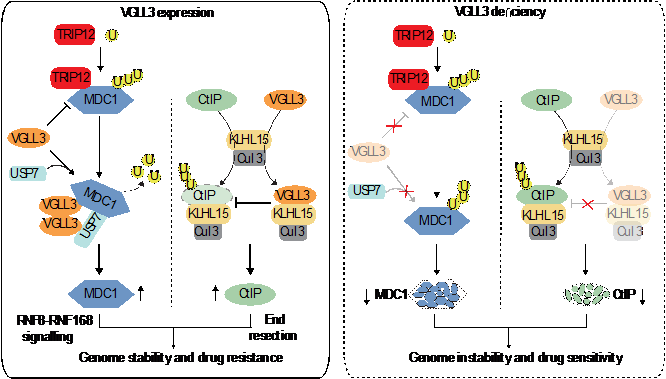
Model for VGLL3 in DDR and chemoresponse (Image by GUO Caixia's group)