China National Center for Bioinformation Releases Influ-BERT, a Genomic Language Model for Influenza Viruses
Influenza A Virus (IAV) poses a persistent threat to global public health due to its rapid mutation and cross-species transmission risks. Traditional surveillance methods, which rely heavily on predefined reference libraries, struggle to identify low-frequency subtypes or analyze incomplete genome sequences. Furthermore, existing general-purpose AI genomic models fail to capture the complex mutation patterns of the influenza genome, leading to significant blind spots when detecting low-frequency subtype crucial for pandemic early warning.
To address this critical gap, a research team led by Prof. Shuhui Song at the China National Center for Bioinformation, collaborated with Prof. Ana Tereza Ribeiro de Vasconcelos from the National Laboratory for Scientific Computing (LNCC) in Brazil, officially released Influ-BERT, a genomic language model tailored for influenza viruses. Based on the Transformer architecture, the model has been deeply optimized for the genomic characteristics of influenza viruses, providing an efficient and intelligent computational solution for applications such as influenza virus subtype identification and pathogenicity prediction.
The study was published in Briefings in Bioinformatics on April 15.
Trained on a massive corpus of approximately 900,000 viral sequences, Influ-BERT core innovation lies in its two-stage training strategy. By combining a customized viral Byte Pair Encoding (BPE) tokenizer with domain-adaptive pretraining, the model successfully bridges the semantic gap between general genomic model and the unique characteristics of influenza, enabling highly precise genomic modeling.
In performance evaluation, Influ-BERT demonstrates superior representation learning capabilities compared to traditional machine learning algorithms and general genomic large models, achieving automated and accurate identification of low-frequency subtypes. Furthermore, the research team expanded the model' application boundaries, successfully utilizing it for key tasks such as differentiating various respiratory viruses (including SARS-CoV-2, rhinovirus, and respiratory syncytial virus), predicting viral pathogenicity, and identifying functional genes. By introducing sliding window perturbation analysis, the study revealed that Influ-BERT autonomously focuses on biologically significant sites. This demonstrates the model's ability to capture the biological functional constraints of the influenza genome without requiring manual annotation.
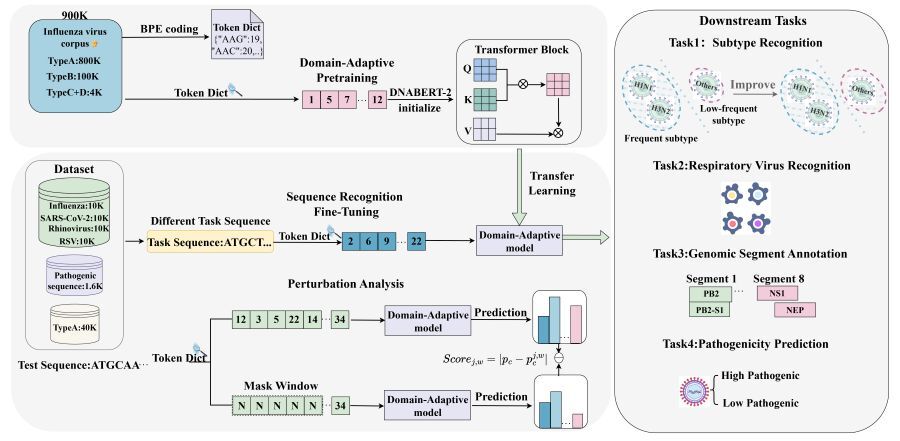
Figure 1: Influ-BERT Workflow Diagram