Transcriptome-wide N6-methyladenosine Profiling of Rice Callus and Leaf Reveals the Presence of Tissue-specific Competitors Involved in Selective mRNA Modification
N6-methyl-adenosine (m6A) is the most abundant modification on the internal messenger RNAs. M6A is regulated by methyltransferase (METTL3, METTL14, and WTAP) / demethylase (FTO, ALKBH5) and some RNA binding proteins (YTHDF1/2/3, ELAVL1), it’s also a dynamic and reversible process. Knockdown or inhibition of m6A enzyme components will lead to important changes related to phenotype, such as Arabidopsis embryonic diapause, human cell apoptosis, yeast gametes developmental defects. Thus, m6A may be a very important intracellular modification.
In 2012, a method of m6A-specific methylated RNA immunoprecipitation combined with next-generation sequencing (MeRIP-seq or m6A-seq) was proposed, which makes large-scale detection of m6A modification possible. Nearly two years, studies of human, mouse, yeast and Arabidopsis in the system, have made great progresses in research on dynamic regulation mechanism of methylation, found that m6A may directly or indirectly affect mRNA nuclear export, translation and degradation. However, Scientists from Beijing Institute of Genomics, Chinese Academy of Sciences found that not all the transcribed mRNAs will be methylated by m6A, but selective methylation for a specific tissue or cell line did exists. They defined the genes which express in two tissues (or cell lines) but only methylated in one tissue (cell line) as Selectively Methylated Genes (SMGs).
In 2013, they first designed and developed the bioinformatics software MeRIP-PF, which can identify m6A peak fast and accurately. Recently, they completed the construction of m6A modified profile of rice callus and leaf tissue using the software and explored the possible mechanism of selectively methylated genes.
This study was the first to reveal the basic characteristics of rice m6A modified spectrum. There are 2~3 m6A modified sites in each mRNA averagely, and the modification of m6A is mainly distributed in the CDS and 3 'UTR region, which is similar to the animal distribution.
Moreover, they found that m6A modification was highly concentrated in the telomeric region of chromosome, and m6A concentration was positively correlated with the number of exon, and the length of intron and whole gene.
At the same time, they also identified SMGs in the callus and leaf tissue, and there are 626 and 5509 genes, respectively. Further analysis of the peak of SMGs revealed that there are binding motifs of other RNA binding proteins (RBPs) in the vicinity of modification sites of m6A. For example, they identified the binding site of PUM gene (UGUA[AC][AC] E-value:8.6e-012)in the SMGs of leaf tissue and found that the 7 genes encoding PUM proteins in leaf tissues had very low expression or even no expression, but have significantly higher expression in the callus. They therefore demonstrate that some RBPs may be as “competitor”, which is competitive with the methyltransferase, when the methyltransferase combines mRNA, resulting in the SMGs in tissue or cell line.
The results not only expand our knowledge on the modification and function of RNA, but also provide an important resource for plant epigenetic transcriptome studies.
This work was supported by National Natural Science.
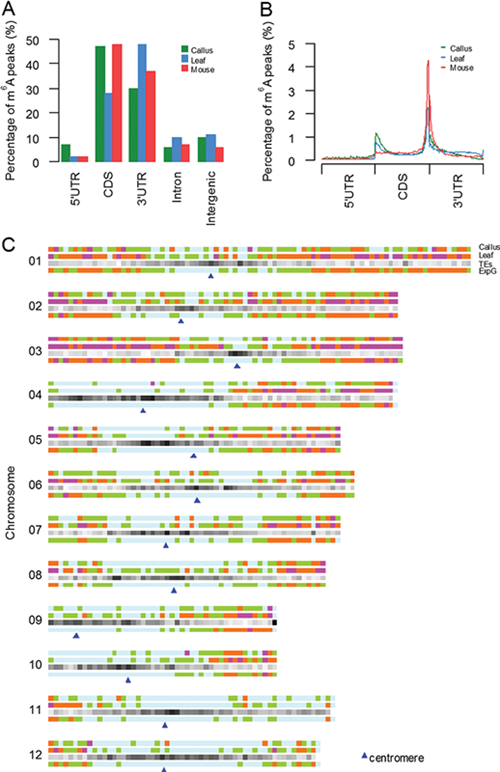
Distribution of m6A modification peaks along mRNA and chromosome (Image by Song Shuhui's group)