China National Center for Bioinformation developed scHILL, an AI-driven tool for deciphering immune heterogeneity at the individual level
In recent years, single-cell foundation models based on large language models have rapidly emerged. These foundation models have demonstrated outstanding performance in downstream tasks such as batch integration, cell type annotation, and regulatory network prediction. However, existing methods mainly focus on identifying cell types and cell states, and have limitation in their ability to capture and quantify heterogeneity at the individual level. Such individual-level heterogeneity is a key factor associated with disease manifestation, therapeutic response, and prognosis, and is particularly important in the study of autoimmune disease, cancer, and infectious disease.
To address this challenge, China National Center for Bioinformation developed scHILL (scRNA-seq data for deciphering Heterogeneity at the Individual-LeveL), a deep learning framework that takes individual single-cell expression matrices as inputs and outputs scores for each individual through a combination of Masked Autoencoder (MAE) and Multilayer Perceptron (MLP). Based on the individual scores, scHILL can perform downstream tasks including phenotypic label prediction, finer patient stratification, and disease-associated genes identification.
In terms of model architecture, scHILL innovatively adopts the Vision Transformer as its backbone model, leveraging its capability of global feature extraction to learn latent dependencies within single-cell expression matrices. Regarding the training strategy, to address the limited number of individual-level single-cell transcriptomic samples in specific disease settings, scHILL randomly crops single-cell expression matrices into multiple smaller matrices. This substantially increases the size of the training dataset, reduces the risk of overfitting, and improves model generalization. In addition, during the pre-training stage, scHILL employs a mask and reconstruction strategy, in which the model reconstructs the remaining 60% masked information using only 40% visible information. This enables the model to learn latent patterns within expression matrices without requiring cell type annotations or phenotype labels.
Researchers validated the effectiveness and interpretability of scHILL using single-cell transcriptomic datasets from patients with infectious diseases, autoimmune diseases, and cancers. First, across multiple COVID-19 peripheral blood mononuclear cell datasets, including datasets not used during pre-training, scHILL outperformed existing models in disease severity prediction. Moreover, scHILL successfully identified patient subgroups beyond conventional clinical classifications. Mild patients with high scHILL scores showed B-cell expansion features resembling those of severe patients, whereas severe patients with low scHILL scores retained CD8⁺ T-cell proportions and cytotoxicity similar to mild patients, demonstrating the potential of scHILL for precision diagnosis and treatment of diseases. Second, in juvenile dermatomyositis peripheral blood mononuclear cell dataset, scHILL scores showed a significant negative correlation with NK cell proportions, consistent with clinical scores assigned by physicians based on patient symptoms. Regression and correlation analyses further revealed that PDE3B was closely associated with disease progression and was highly expressed in naive T cells from patients. Notably, inhibitors targeting this molecular target have already been applied in the clinical treatment of cardiovascular diseases, providing a new perspective for the clinical treatment of juvenile dermatomyositis. Third, in a B cell dataset from multi-organ normal adjacent tissues, scHILL identified two distinct subgroups without fine-tuning, with only one subgroup showing plasma cell enrichment and continuous B cell to plasma cell differentiation. This finding could not be obtained using traditional clustering methods, demonstrating that scHILL can uncover latent individual-level immune state differences in the absence of prior label information.
Overall, scHILL highlights the potential of visual models in understanding single-cell transcriptomic data, and provides a new tool for deciphering individual-level heterogeneity, as well as important methodological support for clinical evaluation.
The study, entitled “scHILL: deciphering individual-level immune cell heterogeneity with single-cell RNA sequencing data,” was published in the international journal Briefings in Bioinformatics. Professor Shuhui Song from the China National Center for Bioinformation is the corresponding author, and Ph.D. candidate Yi Wang is the first author. This work was supported by the Major Research Program “The Immunity Deciphering Project” of the National Natural Science Foundation of China.
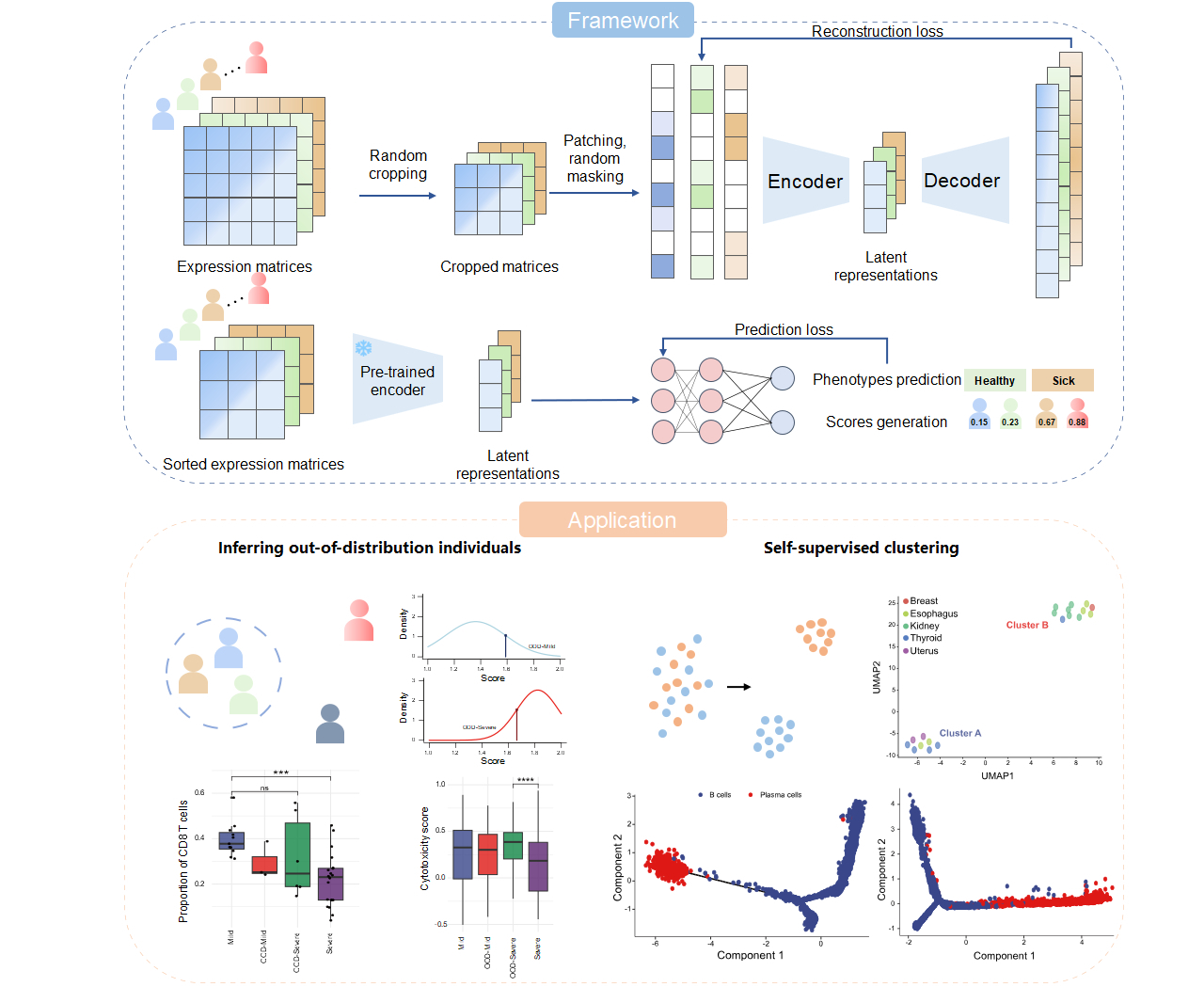
Fig. Architecture and application scenarios of scHILL